Alignment
Beim Alignment werden zwei (paarweises Alignment) oder mehrere Sequenzen (multiples Alignment) miteinander verglichen. Dabei werden die Elemente der einen Sequenz mit denen der anderen so angeordnet, dass die Reihenfolge erhalten bleibt und die einzelnen Elemente zueinander passen bzw. einem Gap (Leerstelle) zugeordnet sind. Die Sequenzen sollten möglichst identisch oder ähnlich sein, um ein alignen überhaupt zu ermöglichen. Fehlpaarungen weisen auf eine Mutation hin, Gaps auf eine Insertion oder Desertion. Große Ähnlichkeiten der Sequenzen können eine Verwandtschaft aufzeigen.
ClustalW (oder ClustalX) ist ein Programm für Multiples Sequenzalignment von Nucleotid- oder Aminosäuresequenzen. ClustalW ist ein Kommandozeilenprogramm, während ClustalX eine graphische Benutzeroberfläche hat. Im Folgenden wird ClustalX vorgestellt. Die Sequenzen können im txt- oder Fasta-Format geöffnet werden.
Zunächst wird paarweise aligniert, worauf ein phylogenetischer Baum erstellt wird. Dieser wird dann wiederum für das multiple Alignment verwendet.
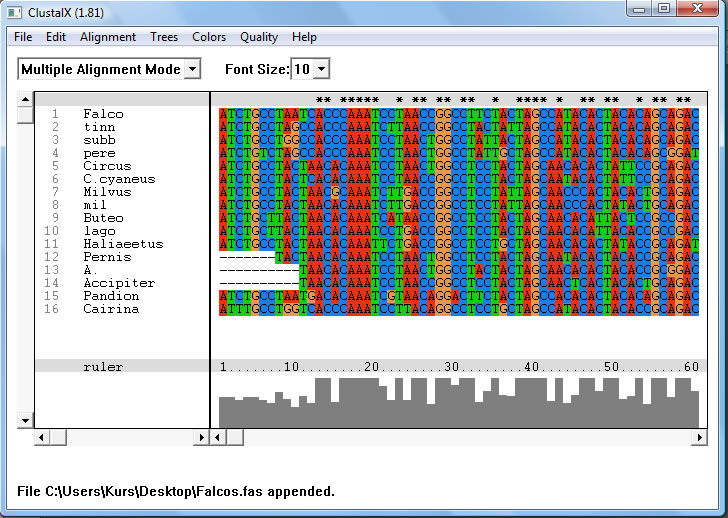
Achtung: Die Taxa müssen alle unterschiedlich heißen, sonst kommt es zu einer Fehlermeldung!
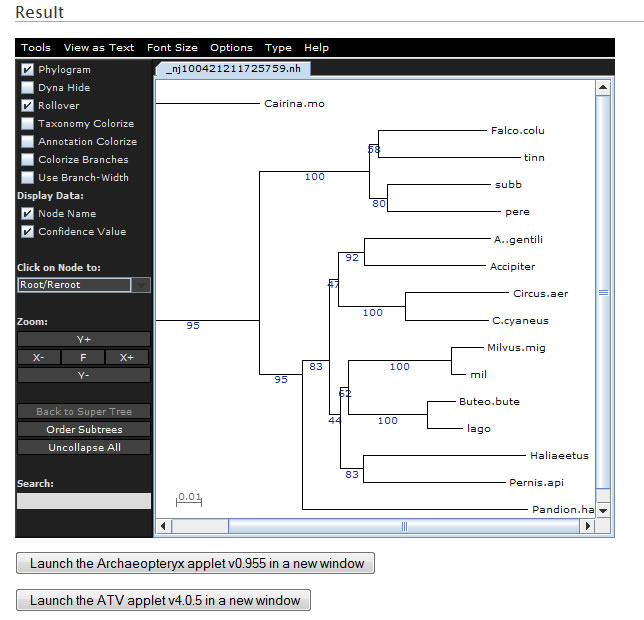
Es kann auch ein phylogenetischer Baum (auch mit Bootstraps) im phb-Format erstellt werden. Dieser kann dann beispielsweise mit TreeView angeschaut werden.
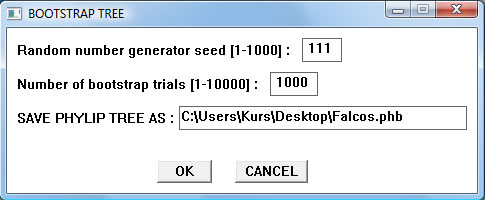
Es ist auch möglich, das multiple Alignment anhand eines sogenannten Guide Trees schrittweise entlang dieses Baumes zu bestimmen. Oder es kann auch nur ein Guide Tree erstellt werden, ohne das komplette Alignment durchzuführen.
Ein Online-Programm zum schnellen Alignen von Nucleotid- oder Aminosäuresequenzen. Die Sequenzen werden entweder direkt eingegeben oder können als Datei im Fasta-Format hochgeladen werden. Das Programm erstellt auch einen phylogenetischen Baum (mit oder ohne Bootstraps). Die alignierten Sequenzen können in verschiedenen Formaten ausgegeben werden, darunter Fasta, Clustal, Nexus und Phylip.
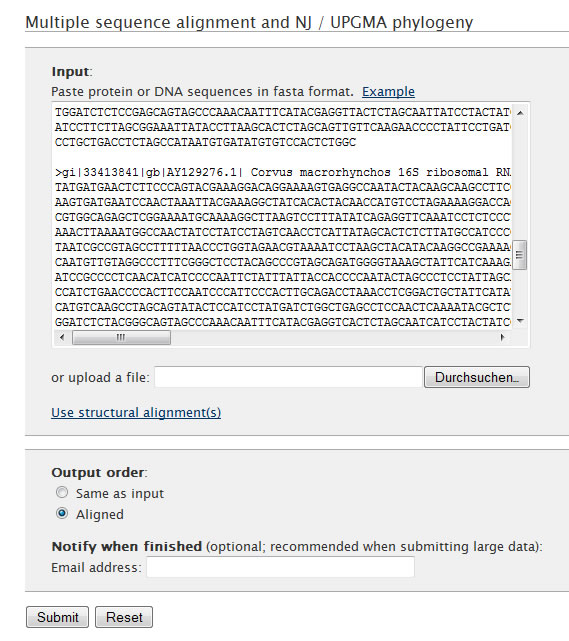
Es ist auch möglich sich per E-Mail informieren zu lassen, wenn das Programm fertig ist. Dies ist bei größeren Datenmengen hilfreich. Weiterhin können verschiedene Methoden des Alignments verwendet werden, je nach Größe des Datensatzes.

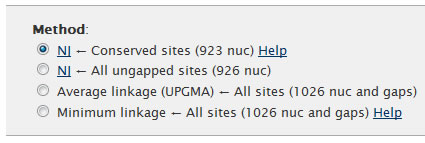
Der phylogenetische Baum kann nach verschiedenen Verfahren erstellt werden: Neighbor-Joining, UPGMA oder Minimum linkage.
Anzusehen ist der Baum dann mit ATV oder Archaeopteryx (benötigt JAVA!). Die Außengruppe kann durch einen Klick auf diese bestimmt werden.